Fenylketonuri
Marian Grosser studerade humanmedicin i München. Dessutom vågade läkaren, som var intresserad av många saker, att göra några spännande omvägar: studera filosofi och konsthistoria, jobba på radion och slutligen också för en Netdoctor.
Mer om -experterna Allt -innehåll kontrolleras av medicinska journalister.Fenylketonuri (PKU) är en medfödd, ärftlig sjukdom av proteinmetabolism. Det förhindrar nedbrytning av aminosyran fenylalanin. Detta ackumuleras i kroppen och stör utvecklingen av barnets hjärna. Om det inte behandlas leder fenylketonuri till allvarliga intellektuella funktionshinder. Med rätt behandling kan patienterna leva ett normalt liv. Ta reda på allt om fenylketonuri här!
ICD -koder för denna sjukdom: ICD -koder är internationellt erkända koder för medicinska diagnoser. De finns till exempel i läkarbrev eller på intyg om arbetsoförmåga. E70
Fenylketonuri: beskrivning
Fenylketonuri (PKU) är en ärftlig metabolisk sjukdom som finns från födseln och stör nedbrytningen av den essentiella aminosyran fenylalanin. Aminosyror är de grundläggande byggstenarna i proteiner och därmed vitala metaboliska komponenter. Några av dem kan bara komma in i kroppen genom mat, organismen kan inte producera dem själv. Sådana aminosyror kallas väsentliga.
Vad händer med fenylketonuri?
Normalt är aminosyror föremål för en balans mellan absorption / uppbyggnad och nedbrytning, så att det alltid finns så mycket tillgängligt som kroppen behöver. När det gäller olika aminosyror kan brist eller överskott orsaka betydande skada och orsaka olika symptom.
I den så kallade klassiska PKU är effekten av fenylalaninhydroxylas (PAH) begränsad eller till och med helt frånvarande. På grund av PAH -bristen ackumuleras fenylalanin alltmer i kroppen. För hög koncentration av fenylalanin stör hjärnans utveckling avsevärt och leder till intellektuella funktionsnedsättningar hos unga patienter i ett tidigt skede.
Eftersom den normala nedbrytningen av fenylalanin inte är möjlig med sjukdomen, bildas andra nedbrytningsprodukter, de så kallade fenylketonerna. De utsöndras i urinen och är ansvariga för namnet på sjukdomen.
Atypisk fenylketonuri
Även med atypiska former av fenylketonuri störs nedbrytningen av fenylalanin. Orsaken är dock inte ett fel på PAH. Istället är funktionen hos ett koenzym, tetrahydrobiopterin (BH4), begränsad. Det är indirekt inblandat i nedbrytningen av fenylalanin eftersom PAH behöver BH4 för att omvandla fenylalanin till tyrosin.
Eftersom BH4 också är viktigt för produktionen av budbärarämnena dopamin och serotonin, är atypisk fenylketonuri med BH4 -brist vanligtvis mer komplicerad än den klassiska formen.
Vem påverkar fenylketonuri?
PKU är en av de vanligaste medfödda metabola sjukdomarna. Det beräknas att cirka en av 7000 nyfödda världen över kommer att utveckla det, utan skillnad mellan flickor och pojkar. Eftersom det är en ärftlig sjukdom påverkas ofta flera i en familj.
Fenyketonuri: symtom
Till en början visar spädbarn med fenylketonuri inga symptom på sjukdomen. De första problemen uppstår inte förrän i fjärde till sjätte månaden i livet, om sjukdomen ännu inte har identifierats och behandlats. Framför allt orsakar den störda hjärnmognaden massiva komplikationer över tiden. Symtom på obehandlad PKU inkluderar:
- ett starkt psykiskt underskott. Hjärnskadan fortskrider genom puberteten och stagnerar sedan. De drabbade barnen är då vanligtvis psykiskt funktionshindrade.
- Passar (anfall) (epilepsi). På grund av skadan är hjärnans nervceller särskilt känsliga och överspännande. Frekventa epileptiska anfall är resultatet.
- motoriska funktionshinder. Inte bara hjärncellerna, utan också patientens muskler kan vara överspända. Det är därför det ofta blir spänt (spasticitet), vilket leder till olika rörelsestörningar.
- Beteendestörningar. Vissa barn med fenylketonuri är hyperaktiva och ovanligt aggressiva, och utbrott av ilska är också vanligare.
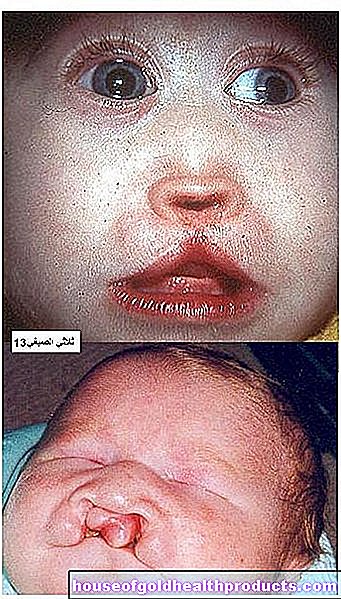
- ett litet huvud (mikrocefali). Eftersom patientens hjärna inte utvecklas ordentligt, faller också huvudtillväxten efter. Den lilla huvudomkretsen jämfört med sina kamrater är särskilt märkbar hos äldre barn.
- en märkbar lukt. PKU producerar vissa nedbrytningsprodukter av fenylalanin som luktar liknande musekskrement. Dessa ämnen utsöndras huvudsakligen i urinen, men också delvis genom huden.
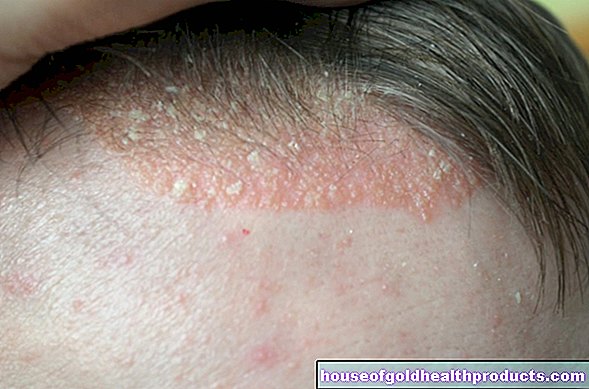
- eksemliknande hudförändringar
Eftersom produktionen av pigmentet melanin också störs i fenylketonuri, har många drabbade mycket ljus, solkänslig hud och vitblont hår. Ögonets iris är också ljusblå till transparent och låter den rödaktiga fundusen lysa igenom.
PKU -symptomen varierar i svårighetsgrad från person till person. Huvudorsaken till detta är att aktiviteten hos fenylalaninhydroxylas (PAH) är begränsad på olika sätt hos varje patient. Vissa har fortfarande en viss kvarvarande aktivitet, så att mindre fenylalanin ackumuleras i organismen. Andra visar ingen enzymaktivitet alls - sjukdomen utvecklas på motsvarande sätt snabbare och mer allvarligt.
Fenylketonuri: orsaker och riskfaktorer
Fenylketonuri är en ärftlig sjukdom. Många genetiska mutationer är nu kända som leder till en defekt i PAH. Typen av mutation avgör i vilken utsträckning nedbrytningen av fenylalanin är begränsad.
PKU ärvs recessivt, vilket innebär att en person kan vara bärare av en förändrad gen utan att utveckla sjukdomen. På samma sätt kan personer med fenylketonuri skaffa friska barn.
Endast om båda föräldrarna har mutationer i sitt genetiska smink är det en viss sannolikhet att deras avkomma kommer att utveckla fenylketonuri. Om föräldrarna inte bara är bärare av generna utan också har PKU själva, kommer alla barn tillsammans att utveckla det.
Fenylketonuri: undersökningar och diagnos
Eftersom de allvarliga konsekvenserna av fenylketonuri kan förebyggas genom att starta behandlingen i god tid, är det särskilt viktigt att upptäcka sjukdomen så tidigt som möjligt. I Tyskland undersöks barn för olika medfödda sjukdomar, inklusive PKU, som en del av en allmän undersökning (nyfödd screening) den tredje dagen efter födseln.
Tandem masspektrometri
Många medfödda metaboliska störningar diagnostiseras nu med hjälp av så kallad tandem-masspektrometri. Det gör att det nyfödda blodet kan undersökas snabbt och enkelt. Förutom fenylketonuri kan läkare upptäcka mer än 20 andra sjukdomar inom några minuter.
Guthrie test
Guthrie -testet, uppkallat efter sin uppfinnare, gör det också möjligt att diagnostisera PKU. För att göra detta tas en liten mängd blod från barnets häl och appliceras på en bit filterpapper. I laboratoriet kan du sedan avgöra om koncentrationen av fenylalanin ökar.
Guthrie -testet introducerades på 1960 -talet och har länge varit standardmetoden för att diagnostisera fenylketonuri. Det har emellertid nackdelar jämfört med tandem -masspektrometri. Guthrie -testet ger bara ett resultat efter fem dagar, vilket också är benäget för fel. Till exempel förfalskar faktorer som barnets kost eller eventuell antibiotikabehandling resultaten. I vissa länder används Guthrie -testet fortfarande, i Tyskland används det vanligtvis inte längre.
Andra tester
Om den nyfödda screeningen avslöjar misstanke om fenylketonuri följer en ytterligare undersökning för bekräftelse. Detta kan också användas för att bestämma den exakta koncentrationen av fenylalanin i blodet.
Slutligen är det viktigt att skilja på om det är en typisk (klassisk) eller atypisk PKU. Särskilda tester är också tillgängliga för detta, till exempel tetrahydrobiopterin stresstest. Skillnaden är viktig eftersom atypisk fenylketonuri behandlas annorlunda än den klassiska formen.
Fostervattenundersökning
Fenylketonuri kan redan diagnostiseras under graviditeten (prenatal diagnos). För att göra detta tas en liten mängd fostervatten från moderns fostersäck och cellerna hos det ofödda barnet som finns i den undersöks. Eventuella genetiska defekter som orsakar PKU kan identifieras på detta sätt.
Med screening för nyfödda upptäcks dock fenylketonuri vanligtvis tillräckligt tidigt för att behandlas. Eftersom ett fostervattentest alltid är förknippat med en viss risk är dess användning för diagnos av PKU vanligtvis inte användbar.
Fenylketonuri: behandling
Det finns bara ett sätt att motverka överskott av fenylalanin i PKU: De drabbade barnen måste följa en särskild kost för att få i sig så lite fenylalanin som möjligt med maten. De behöver också vissa kosttillskott för att ersätta ämnen som skulle bildas av fenylalanin hos friska barn.
Terapin måste påbörjas innan de första symptomen på en utvecklingsstörning uppträder, det vill säga inom de första två månaderna av livet. Hjärnskador som redan har inträffat går inte att vända.
Näringsterapi med PKU -kosten
Alla naturliga proteiner består av cirka fem procent av aminosyran fenylalanin. De flesta livsmedel innehåller mycket mer av det än kroppen behöver. Detta är inte ett problem för en frisk person eftersom han bryts ner och utsöndrar de överflödiga mängderna fenylalanin.
Patienter med fenylketonuri, å andra sidan, måste klara sig utan naturliga proteiner för det mesta. Industriellt tillverkade specialprodukter ersätter de saknade livsmedelskomponenterna. Å ena sidan vill man förhindra ett överskott av fenylalanin, å andra sidan bör patienten förses med ämnen som annars skulle saknas, till exempel tyrosin, som bildas av fenylalanin.
Men målet med PKU -kosten är inte att helt stoppa absorptionen av fenylalanin. Eftersom organismen behöver en viss mängd aminosyra för viktiga metaboliska processer. Att uppnå rätt koncentration utgör en stor utmaning för läkare och patienter och kräver mycket disciplin.
Dieten måste därför följas under en livstid, men särskilt strikt fram till sex års ålder. Eftersom hjärnan utvecklas mycket starkt fram till denna ålder och därför är särskilt mottaglig för skador. I vuxen ålder orsakar höga koncentrationer av fenylalanin inte hjärnskador som de gör hos barn. De kan dock vara utlösaren för andra neurologiska besvär såsom dålig koncentration eller saktade reaktioner.
Dietbehandling för fenylketonuri bör börja på ett specialistläkare för metabolisk sjukdom. För det är inte möjligt på varje klinik att instruera föräldrar om kosten. Detta görs bäst med hjälp av kostrådgivning, som visar hur du går till kosten och regelbundet övervakar blodfenylalaninnivåer.
Behandling av atypisk fenylketonuri
I atypiska former av PKU ersätts det saknade koenzymet BH4 och vissa budbärarsubstanser som dopamin och serotonin artificiellt. I vissa fall måste patienten också ha en lågfenylalanindiet.
Fenylketonuri under graviditeten
Om du är gravid och själv har fenylketonuri bör du överväga följande:
- Håll dig till din diet särskilt strikt!
- Låt dina fenylalaninblodvärden kontrolleras med jämna mellanrum. Eftersom koncentrationerna av fenylalanin hos det ofödda barnet är ungefär dubbelt så höga som hos den gravida kvinnan. Förutom hjärnan kan de också skada hjärtat och ögonen på det ofödda barnet. Missbildningar av barnets skelett kan också uppstå från höga fenylalaninnivåer under graviditeten.
- För att utesluta hjärnskador på barnet i början av graviditeten bör kvinnor med fenylketonuri planera graviditeten noggrant och vidta försiktighetsåtgärder från början.
- Hos gravida kvinnor med fenylketonuri kan en diet som inte strikt följs leda till missfall (spontan abort).
- I alla fall bör gravida kvinnor med fenylketonuri söka råd och vägledning från en läkare.
Fenylketonuri: sjukdomsförlopp och prognos
Om fenylketonuri diagnostiseras tidigt, om möjligt hos den nyfödda, och om den speciella PKU -kosten följs, är prognosen vanligtvis bra. Barnen utvecklas andligt och har en genomsnittlig livslängd.
Om den inte behandlas leder hjärnskadan till allvarliga psykiska utvecklingsstörningar som inte kan korrigeras senare. De drabbade har en normal livslängd, men deras intelligenskvot är nästan alltid mycket långt under normen.
Den sällsynta atypiska formen av fenylketonuri, där BH4 -brist förekommer, är ett speciellt fall. Denna variant av fenylketonuri kan leda till progressiv neurologisk skada med svåra kramper trots kost.
Tagg: diet hudvård läkemedel







-bei-kindern.jpg)