Angelmans syndrom
Lisa Vogel studerade institutionell journalistik med fokus på medicin och biovetenskap vid Ansbach University och fördjupade sina journalistiska kunskaper inom magisterexamen i multimediainformation och kommunikation. Detta följdes av en praktikplats i -redaktionen. Sedan september 2020 har hon skrivit som frilansjournalist för
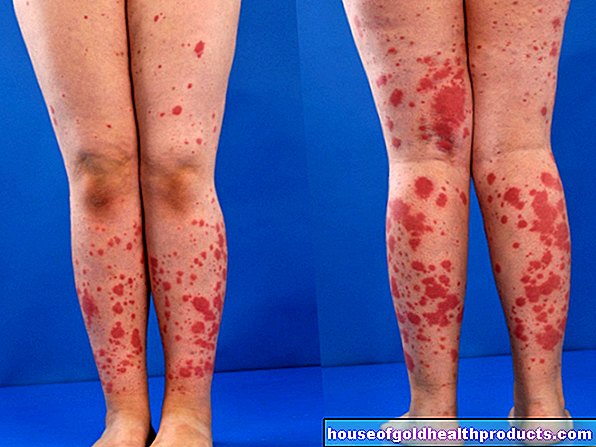
Fler inlägg av Lisa Vogel Allt -innehåll kontrolleras av medicinska journalister.Angelmans syndrom (Happy Puppet Syndrome) är en sällsynt, genetisk sjukdom. Det manifesterar sig bland annat genom mentala och fysiska begränsningar, utvecklingsstörningar (särskilt språk) och hyperaktivitet. Det som är slående är det dockliknande utseendet och de drabbades glada ansiktsuttryck. Läs mer om den sällsynta sjukdomen!
ICD -koder för denna sjukdom: ICD -koder är internationellt erkända koder för medicinska diagnoser. De finns till exempel i läkarbrev eller på intyg om arbetsoförmåga. Q93

Kort överblick
- Vad är Angelmans syndrom? Sällsynt, genetisk sjukdom som manifesterar sig genom mentala och fysiska begränsningar i barns utveckling
- Symtom: dockliknande ansiktsdrag, utvecklingsstörning, försämrad koordination, ingen eller knappast någon språkutveckling, minskad intelligens, anfall, grundlöst skratt, skrattanfall, överdriven dregling, glädjande viftning av armarna
- Orsaker: genetisk defekt på kromosom 15
- Diagnos: bland annat samtal, fysisk undersökning, genetiska undersökningar
- Terapi: Ingen kausal terapi tillgänglig; Stödjande t.ex. sjukgymnastik, logoped, arbetsterapi; eventuellt medicin för att lindra symtom (t.ex. vid anfall)
- Prognos: normal livslängd; inget självständigt liv möjligt
Angelmans syndrom: definition
Angelmans syndrom (AS) orsakas av en genetisk defekt på kromosom 15. Denna defekt stör den drabbade fysiska och mentala utvecklingen. Talutvecklingsstörningar, motorisk osäkerhet och ett lyckligt ansikte är de mest märkbara symtomen på Angelmans syndrom.
Namnet "Angelmans syndrom" kommer från upptäckaren av sjukdomen, den engelska barnläkaren Harry Angelman. 1965 jämförde han de kliniska bilderna på tre barn som hade dockliknande ansiktsdrag. Barnen skrattade mycket och gjorde ryckiga rörelser - som marionetter Detta ledde till det engelska namnet "Happy Puppet Syndrome" (happy puppet).
Angelmans syndrom förekommer hos båda könen. Risken för sjukdomen är cirka 1: 20000. Detta gör syndromet till en sällsynt sjukdom.
Angelmans syndrom: symtom
Vid födseln är barn med Angelmans syndrom normala. Motoriska och kognitiva utvecklingsstörningar blir bara alltmer märkbara i spädbarn och tidig barndom. Funktioner hos den genetiska störningen är:
- försenad motorisk utveckling
- försämrad koordination
- ofta ingen eller knappast någon språkutveckling
- minskad intelligens
- hyperaktivt, högljudd beteende
- grundlöst skratt
- Passar skratt
- glada gester (t.ex. vifta med armarna)
- överdriven dregling
- sticker ofta ut ur tungan
Vissa barn med Angelmans syndrom har också:
- Mikrocefali (onormalt litet huvud) - inte vid födseln, men som det utvecklas
- Beslag
- Förändringar i elektrisk hjärnaktivitet
- mycket ljus hud och ögon på grund av minskad pigmentering (hypopigmentering)
- Squint (strabismus)
Angelmans syndrom: orsaker
Orsaken till Angelmans syndrom är en genetisk defekt på kromosom 15: UBE3A -genens funktion försämras hos de drabbade. Denna gen producerar normalt ett enzym som är involverat i att bryta ner skadade eller överflödiga proteiner i cellerna. Det hjälper alltså cellen att fungera normalt.
UBE3A -genen ligger i kromosomregionen 15q11q13. Där utsätts generna för det som kallas "genomisk prägling". Det betyder att de bara är aktiva på en av föräldrakromosomerna (i våra kroppsceller finns det två kopior av varje kromosom - en från modern och en från fadern). Detta regleras av en kemisk process - metylering: metylgrupper fästa vid vissa punkter kan slå på eller av en gen.
Genen är aktiv på båda kromosomerna i många kroppsceller, men inte i hjärnans nervceller: hos många människor stängs UBE3A -genen av på faderlig kromosom 15 genom avtryck. Som ett resultat är UBE3A endast aktivt på maternell kromosom 15 i hjärnan. Detta betyder också: Om den moderna genkopian visar ett fel kan detta inte kompenseras av den nedlagda fadergenkopian. Och det är just denna kombination som förekommer i Angelmans syndrom: faderns gensegment är avstängt, moderns gensegment är defekt.
Det underliggande genetiska felet kan vara av olika typer:
- Radering: Cirka 75 procent av alla personer med Angelmans syndrom saknar den relevanta regionen 15q11q13 med UBE3A -genen på maternell kromosom 15. Eftersom motsvarande region på faderlig kromosom 15 "stängs av" genom prägling kan kroppen använda enzymet vars konstruktionsplan sparas i Gen UBE3A, skapa inte.
- Mutation i UBE3A -genen: En spontant förändring i genen gör att informationen i den går förlorad. Detta gäller för fem till tio procent av personer med Angelmans syndrom. I ungefär vart femte fall finns det en familjär mutation: Här bär modern redan den genetiska förändringen på sin fars kromosom.
- Två faderliga kromosomer 15: Den drabbade personen ärvde båda kromosomerna 15 från sin far, ingen från modern (medicinskt kallad "paternal uniparental disomy 15"). Så det finns ingen aktiv UBE3A -gen. Detta gäller cirka en till två procent av alla patienter med Angelman syndrom.
- Inskriftsfel: UBE3A -genen på maternell kromosom 15 - som den på paternal kromosom 15 - stängs av genom imprinting. Dessutom kan en viss del av kromosomen saknas (radering). Ett tryckfel finns i en till fyra procent av fallen av Angelman syndrom.
I de återstående tio till 15 procent av fallen är orsaken till Angelmans syndrom okänd. Förresten: om modergenen stängs av och faderns gen är defekt, lider barnen av det så kallade Prader-Willis syndrom.
Är Angelmans syndrom ärftligt?
I allmänhet är risken för återfall i Angelmans syndrom låg. Detta innebär risken att föräldrar till ett drabbat barn får andra barn som också har syndromet. I enskilda fall beror denna risk dock till stor del på den genetiska defekt som ligger till grund för Angelmans syndrom.
Till exempel, vid Angelmans syndrom på grund av två faderliga kromosomer 15 (faderlig uniparental disomi 15) är det mindre än en procent. Å andra sidan kan Angelmans syndrom på grund av ett tryckfel med förlust av ett visst gensegment (IC -deletion) inträffa i hälften av alla fall hos ett syskon.
Denna ökade risk finns också med en UBE3A -mutation - förutsatt att modern redan är bärare av den genetiska defekten (i cirka 20 procent av alla mutationsfall). I sådana fall ärvde mamman mutationen från sin far. UBE3A ändras därför i moderns faderliga kromosom. Om detta är avstängt kommer modern inte att märka mutationen. Men hon kan ge kromosomen vidare till sina barn, där den - då som moderkromosomen - kan orsaka Angelmans syndrom.
Teoretiskt kan patienter med Angelmans syndrom reproducera sig. Beroende på när kausala kromosomförändringar inträffade (t.ex. redan under könscellsutveckling eller omedelbart efter befruktning) är risken ibland mycket hög (upp till 100 procent) att de drabbade kommer att överföra sjukdomen. Det saknas dock tillförlitliga uppgifter om detta. Det fanns ett isolerat fall i september 1999: Mamman, som hade Angelmans syndrom, överförde sjukdomen här.
Angelmans syndrom: diagnos
Om du märker symtomen som beskrivs ovan hos ditt barn är barnläkaren den första kontaktpunkten. Han kan begränsa de möjliga orsakerna mer exakt och vid behov hänvisa dig och ditt barn till en specialist.
anamnese
Det första steget i diagnosen är en grundlig medicinsk historia. Läkaren kommer att ställa dig olika frågor om ditt barn, till exempel:
- Vilka förändringar märkte du hos ditt barn?
- Har ditt barn några fysiska klagomål?
- Kan ditt barn sitta?
- Räcker ditt barn efter föremål?
- Talar ditt barn?
- Är ditt barn ofta märkbart glad eller upphetsad?
- Skrattar ditt barn i olämpliga situationer, till exempel när de har ont?
Fysisk undersökning
Därefter följer den fysiska undersökningen. Barnläkaren testar i vilken utsträckning barnet regelbundet utvecklar motoriska och mentala färdigheter. Enkla övningar används för detta ändamål: Till exempel bör barnet koncentrera sig på leksaker eller selektivt nå en byggsten. Läkaren uppmärksammar också barnets ansiktsuttryck. Ofta skratt, dockliknande funktioner och dregla är alla tecken på Angelmans syndrom.
Om det efter den fysiska undersökningen finns misstanke om den sällsynta sjukdomen kommer läkaren att hänvisa dig till en neurolog och en humangenetiker.
Genetiska tester
Genetisk testning är en viktig del av diagnosen Angelmans syndrom. Läkaren behöver ett litet prov av barnceller, som han kan få från munslemhinnan, till exempel genom att ta ett blodprov eller genom att ta en pinne. Det genetiska materialet (DNA) för dessa celler eller den relevanta kromosomregionen 15q11q13 undersöks sedan mer detaljerat i laboratoriet.
I ett första steg uppmärksammar läkarna kromosomsegmentets metyleringsmönster (metyleringsanalys / test). Ytterligare tester på samma prover (deletionsanalys, mutationsanalys) hjälper till att fastställa orsaken till Angelmans syndrom närmare. För detta kan det också vara nödvändigt att undersöka föräldrarnas genetiska sammansättning. På detta sätt kan läkare avgöra om det redan finns en genetisk defekt där.
Ytterligare utredningar
Ytterligare undersökningar är ofta användbara. Till exempel kan EEG användas för att upptäcka förändringar i elektrisk hjärnaktivitet, vilket ofta förekommer i Angelmans syndrom. Oftalmologiska undersökningar kan också anges.
Angelmans syndrom: terapi
Angelmans syndrom är obotligt - den genetiska orsaken till sjukdomen kan inte åtgärdas. Tidigt ingripande kan dock ha en positiv effekt på de drabbades motoriska och mentala utveckling. Regelbunden sjukgymnastik, till exempel, är till hjälp. Det kan förbättra barns motoriska färdigheter, lindra begränsad rörlighet och hjälpa till att förhindra sekundära sjukdomar som ryggmärgsdeformiteter. Barn med Angelmans syndrom har också nytta av andra terapimetoder som arbetsterapi och logoped.
Dessutom kan vissa symptom och tillstånd i samband med Angelmans syndrom kräva målinriktad behandling. Till exempel hjälper antikonvulsiva läkemedel (antiepileptika) mot anfall, och lugnande medel (lugnande medel) hjälper till med allvarliga sömnstörningar.
På webbplatsen för Verein Angelman e.V. hittar du mycket information om Angelmans syndrom, erfarenhetsrapporter och händelser för de drabbade samt kontaktpersoner för de drabbade i alla regioner i Tyskland.
Angelmans syndrom: sjukdomsförlopp och prognos
Första året av livet
Spädbarn med Angelmans syndrom har större risk att amma, suga och svälja. De sticker ofta ut tungorna eller dreglar mycket. Dessutom spottar barn med Angelmans syndrom ofta (vilket ofta misstas som matintolerans eller refluxsjukdom). Ofta kräkningar kan leda till farlig viktminskning.
Vid tre till sex månaders ålder börjar barn med Angelmans syndrom ofta le. De fnissar och gurglar mycket och sticker ofta ut tungan under dessa glädjeutbrott.
Den försenade motoriska utvecklingen märks vanligtvis mellan 6 och 12 månader: barnen kryper inte eller sitter. Överkroppens rörelser är ofta skakiga. Detta i sin tur gör det svårare att sitta.
En bråkdel av de drabbade får anfall redan vid 12 månaders ålder.
Ett till tre år
Inom de första tre åren av livet blir utvecklingsstörningen vid Angelmans syndrom mycket tydlig. Barnen lider mer av lindriga anfall. Några av dem är hyperaktiva, överspända och alltid i rörelse. Många har en tendens att stoppa händerna eller leksakerna i munnen hela tiden, eller att sticka ut tungan ofta och dregla. Om barnen är särskilt upphetsade kommer de ofta att skratta överdrivet och vinka med utsträckta armar.
Den nedsatta språkutvecklingen blir uppenbar för första gången i denna ålder. Barn "babblar" för sig själva eller skriker och gnisslar, men kan bara uttala några få eller inga lättbegripliga ord och brukar använda dem utan sammanhang.Men de förstår språk och det finns också social interaktion med andra människor.
Puberteten och vuxenlivet
Puberteten är ofta tre till fem år försenad hos barn med Angelmans syndrom. Men sexuell mognad utvecklas då normalt. Det finns fortfarande ingen språkutveckling om språkförståelse ofta är närvarande. Anfall i vuxen ålder kan vanligtvis hanteras bra med medicinering.
Personer med Angelmans syndrom har en normal livslängd. Ett självständigt liv är inte möjligt på grund av de mentala begränsningarna.
Tagg: tidskrift klimakteriet terapier







.jpg)