Cystisk fibros
och Christiane Fux, medicinsk redaktör och Martina Feichter, medicinsk redaktör och biologSophie Matzik är frilansskribent för medicinska team.
Mer om -experternaChristiane Fux studerade journalistik och psykologi i Hamburg. Den erfarna medicinska redaktören har skrivit tidningsartiklar, nyheter och faktatexter om alla tänkbara hälsoteman sedan 2001. Förutom sitt arbete för är Christiane Fux också aktiv i prosa. Hennes första kriminalroman publicerades 2012, och hon skriver, designar och publicerar också sina egna kriminalpjäser.
Fler inlägg av Christiane FuxMartina Feichter studerade biologi med ett valbart ämne apotek i Innsbruck och fördjupade sig också i en medicinsk växts värld. Därifrån var det inte långt till andra medicinska ämnen som fortfarande fängslar henne till denna dag. Hon utbildade sig till journalist vid Axel Springer Academy i Hamburg och har arbetat för sedan 2007 - först som redaktör och sedan 2012 som frilansskribent.
Mer om -experterna Allt -innehåll kontrolleras av medicinska journalister.
Cystisk fibros är en medfödd metabolisk sjukdom. Hos de drabbade är kroppsvätskor som saliv, bronkialslem eller bukspottkörtelns sekret mycket tuffare än vanligt. Detta orsakar bland annat andningsproblem och matsmältningsbesvär. Det finns inget botemedel mot cystisk fibros. Med konsekvent behandling kan dock sjukdomsförloppet bromsas. Läs här om symptomen på cystisk fibros och hur man behandlar dem.
ICD -koder för denna sjukdom: ICD -koder är internationellt erkända koder för medicinska diagnoser. De finns till exempel i läkarbrev eller på intyg om arbetsoförmåga. E84
Cystisk fibros: snabb referens
- Beskrivning: ärftlig metabolisk sjukdom, orsakar tjockt slem i lungorna och andra organ
- Symtom: andningsproblem, torr hosta, känslighet för lunginfektioner, misslyckande med att trivas, matsmältningsbesvär, svår diarré, fet lever, minskad fertilitet
- Orsaker: nedärvning av defekta gener som påverkar kroppsvätskornas konsistens. Sjukdomen bryter dock bara ut om båda föräldrarna vidarebefordrar en sjuk gen till barnet (autosomalt recessivt arv)
- Diagnostik: blodprov för immunreaktivt trypsin (IRT), pankreatitassocierat protein (PAP), svettprov, genetiskt test
- Behandling: slemförtjockningsmedel, bronkodilatatorer, inandning, antibiotika för bakteriella luftvägsinfektioner, kortison, CFTR-modulatorer, lungtransplantation
- Prognos: obotlig, kursen beror starkt på diagnosens svårighetsgrad och tid, förkortad livslängd
Cystisk fibros: beskrivning
Cystisk fibros (även kallad cystisk fibros) är en ärftlig metabolisk sjukdom. Bildningen av olika kroppsvätskor störs. Utsöndringen av lungorna, bukspottkörteln och andra organ är mer trögflytande än hos friska människor.
Tjockt slem
Det hårda slemet täpper till bland annat bronkiernas små grenar och kanalerna i de inre organen. Andning och matsmältning påverkas särskilt hårt. Under sjukdomsförloppet kan organen arbeta allt mindre.
Detta är vad som händer med cystisk fibros
Fel i genomet
Orsaken till sjukdomen är defekter i det genetiska sminket. Cystisk fibros kan därför inte botas. Diagnostid och svårighetsgrad av symtom kan variera mycket från person till person. Hos många barn märks cystisk fibros från födseln, i andra fall känns det först senare.
Cystisk fibros: symptom
Cystisk fibros symptom kan variera mycket från en patient till en annan. Sjukdomen påverkar funktionen hos olika organ, men särskilt lungorna och matsmältningssystemet.
Känner igen tidiga tecken
När de första symptomen uppträder vid cystisk fibros varierar från person till person. I de flesta fall uppträder symptom på cystisk fibros inom det första levnadsåret. Detta innebär att sjukdomen vanligtvis kan diagnostiseras tidigt och behandlingen kan startas snabbt. Vissa patienter har dock bara tydliga symptom under tonåren. Dessutom visar inte alla personer som påverkas hela spektrumet av möjliga symptom. Svårigheten av symtomen varierar också.
Förändrade kroppsvätskor
Vid cystisk fibros störs bildandet av de så kallade kloridjonkanalerna i cellmembranen. Detta förändrar kroppsvätskornas sammansättning. Det enklaste sättet att bestämma denna förändring i svett hos de drabbade. Deras svett är mer salt än hos friska människor. Salterna natrium och klorid, som ingår i de så kallade elektrolyterna, ackumuleras i din svett. De som lider av cystisk fibros förlorar mer kroppssalter genom svettning.
Flera symptom på cystisk fibros
Sjukdomen påverkar ett antal organsystem. De första symptomen på cystisk fibros uppträder ofta i lungorna och mag -tarmkanalen. Ytterligare klagomål kan uppstå under livets gång. Symtomen kan behandlas väl genom riktad terapi. Symtomen kan dock också vara hotfulla. Det är särskilt farligt när bronkialrören blockeras av det tjocka slemmet. Då kan patienterna i extrema fall kvävas.
-
"Idrott kan lindra symtom"
Tre frågor för
Suat Evcümen,
Specialist i internmedicin och pulmonologi -
1
Varför erkänns cystisk fibros så sent hos vissa människor?
Suat EvcümenCystisk fibros är en genetisk sjukdom. Beroende på hur allvarliga mutationerna är, finns det också milda kurser som först blir märkbara sent. I princip fungerar vissa kloridkanaler inte ordentligt, varför patientens kroppssekret tjocknar. Detta påverkar flera organsystem, såsom luftvägarna eller matsmältningen. Det är därför man talar om en multisystemsjukdom.
-
2
Tipsar du nyfödd screening?
Suat EvcümenTidig upptäckt är oerhört viktigt och kan bidra till att påverka sjukdomsförloppet positivt genom tidig behandling. Dessa inkluderar till exempel administrering av matsmältningsenzymer, inhalationsterapi och tidigt fysioterapeutiskt stöd. Av denna anledning har nyfödd screening gjorts i Tyskland under ett antal år.
-
3
Vad innebär diagnosen?
Suat EvcümenLivslängden för patienter med cystisk fibros har ökat stadigt under de senaste två decennierna. Vi är skyldiga detta till den tidiga upptäckten och den tidiga starten av de individuellt anpassade terapierna. De flesta växer upp med sjukdomen och ”lär sig” att hantera den. Vi fortsätter att observera att träning hjälper och lindrar symtom. Sök också psykologisk hjälp.
-
Suat Evcümen,
Specialist i internmedicin och pulmonologiSedan mars 2019 har Suat Evcümen varit överläkare i pulmonologi vid Paracelsus Clinic på Silbersee.

Cystisk fibros: symptom på lungorna
Andningssvårigheter och torr hosta
I de flesta fall av cystisk fibros uppträder inte lungsymtom förrän något äldre barn. Nyfödda har vanligtvis inga andningsproblem. Hos lite äldre barn har symtomen på cystisk fibros ofta formen av kikhostaliknande, kronisk irritabel hosta. Slem i deras luftvägar är ökat, tjockt och tjockt. Detta hindrar luftflödet i lungorna. Med tiden utvecklas andningssvårigheter.
Frekventa infektioner
Den ökade uppbyggnaden av slem i lungorna gör det lättare för bakterier att bosätta sig och orsaka en infektion. Återkommande lunginflammation eller bronkiala infektioner orsakas huvudsakligen av bakterier som stafylokocker och Pseudomonas -arter. Den störda saltbalansen i lungorna hindrar också kroppens försvar. Lungblödning kan också förekomma. Ett typiskt tecken på detta är att hosta upp slem blandat med blod.
Även om lungorna redan är skadade från tidig barndom, uppträder de första symptomen på cystisk fibros i luftvägarna ofta bara i grundskolealdern eller till och med senare. Symtomen är ibland bara märkbara när stora delar av lungvävnaden redan har förstörts eller luftvägarna är kraftigt förträngda.
Cystisk fibros: bukspottkörtelns symptom
Hos patienter med cystisk fibros blir bukspottkörteln ofta inflammerad: Den utsöndrar en utsöndring som bland annat innehåller enzymer för fett- och kolhydratsmältning (exokrin pankreasfunktion). Hos personer med cystisk fibros byggs sekretet upp på grund av dess viskositet och orsakar inflammation i bukspottkörteln (pankreatit).
När processen fortskrider hårdnar bukspottkörteln och blir ärr. Läkare talar om fibros. Fibros förstör gradvis bukspottkörteln.
Förutom matsmältningsutsöndringen producerar bukspottkörteln också hormonet insulin (endokrin pankreasfunktion) i speciella celler - betacellerna. Bland annat är det nödvändigt för kroppen att använda socker. Hos patienter med cystisk fibros förstör gradvis beta -cellerna det viskösa matsmältningssekretet i bukspottkörtelkanalerna - insulinproduktionen försämras och en insulinbrist utvecklas (särskilt hos patienter över 10 år). Som ett resultat kan en form av diabetes (diabetes mellitus) utvecklas: typ 3 -diabetes.
Cystisk fibros: symptom på galla
Bukspottkörteln och gallblåsan delar en gemensam kanal i tarmen. Därför kan eftersläpningen av pankreassekret också orsaka inflammation i gallblåsan. Gallstenar bildas ofta också, vilket helt kan blockera dränering av gallan från gallblåsan.
Cystisk fibros: symptom på matsmältningskanalen
Förutom klagomål i lungorna orsakar cystisk fibros ofta symtom i matsmältningskanalen. Till exempel påverkar bristen på galla fettsmältningen. Som ett resultat har patienter ofta svårt att tolerera fet mat. Det mesta av den intagna maten utsöndras osmält. Mycket omfattande och mjuk avföring är typisk.
Diarré och hämmad tillväxt
Berörda barn, inklusive spädbarn, lider ofta av svår diarré. Även om de dricker och äter tillräckligt, går de knappt upp i vikt. Stunted tillväxt och undernäring är därför ytterligare klassiska konsekvenser av sjukdomen.
Sådan matsmältningsbesvär kan också förekomma med många andra sjukdomar. Endast i kombination med andningsproblem är de en indikation på cystisk fibros, som du definitivt bör undersöka.
Anal prolaps
När det utvecklas kan cystisk fibros leda till olika komplikationer i matsmältningskanalen. Det vanligaste är det som kallas anal prolaps. En del av anus slemhinnan bular ut ur anus. En sådan incident måste opereras så snart som möjligt.
Tarmobstruktion
En invagination av tarmen (intussusception) eller en intestinal obstruktion (ileus) förekommer också ofta. Båda komplikationerna åtföljs av svår buksmärta och betydande matsmältningsproblem. Smärtan kommer vanligtvis i anfall, särskilt efter att ha ätit. En tarmobstruktion är dödlig om den lämnas obehandlad. Krampaktig, akut buksmärta bör därför alltid klargöras av en läkare.
Cystisk fibros: leversymtom
Fet lever
Levern lider också av eftersläpning av galla. Fettlever utvecklas hos många patienter när sjukdomen fortskrider. Detta kan åtföljas av trötthet, aptitlöshet, en känsla av fullhet och flatulens och, i sällsynta fall, känslor av tryck eller lätt smärta i övre delen av buken.
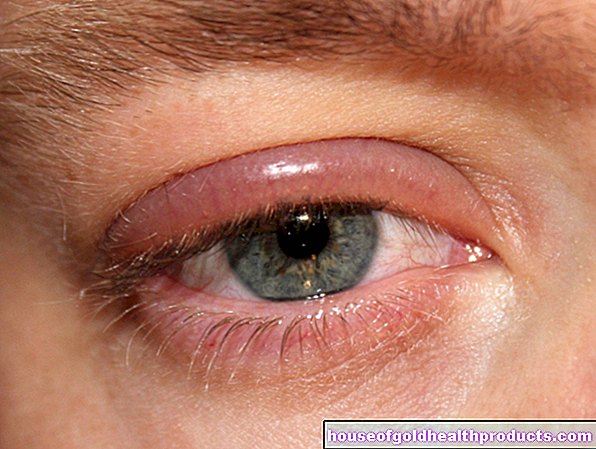
Krymp lever
I sällsynta fall utvecklar patienter med cystisk fibros krympad lever (levercirros). Denna allvarliga leverdysfunktion blir först märkbar i form av gulsot (gulsot). En gulaktig missfärgning av ögonvitorna är ett tecken på gulsot. Hjärtproblem uppstår också efter en lång tid och prestandan hos de drabbade fortsätter att minska.
Cystisk fibros: Minskad fertilitet
Mer än hälften av alla manliga cystisk fibrospatienter är infertila. I de flesta fall producerar de drabbade befruktbara spermier i sina testiklar. Dessa kan dock inte komma igenom vas deferens eftersom de blockeras av visköst slem.
Kvinnor med cystisk fibros är vanligtvis mindre fertila. I princip kan de få barn och bära barn. Dock ackumuleras tufft slem i äggledarna, som spermierna knappt kan tränga igenom. Sannolikheten för graviditet minskar snabbt, särskilt när människor blir äldre.
Cystisk fibros: symptom hos barn
Cystisk fibros är en genetisk sjukdom. Den har därför alltid funnits sedan födseln. Men de klassiska symptomen på cystisk fibros dyker inte alltid upp i barndomen. Det finns dock ofta ospecifika tips som bör följas upp. Detta gäller särskilt om familjen redan har haft fall av cystisk fibros.
Symtom i matsmältningskanalen
En indikation på en metabolisk störning är till exempel en uppblåst mage under lång tid. Barnen lider ofta av diarré. Hos nyfödda kan en mycket fördröjd första tarmrörelse (Kindspech) vara en indikation på cystisk fibros. I många fall uppstår misslyckande med att frodas och stunt trots att barnen äter med ravenous aptit. Förstoppning till följd av sjukdomen förekommer endast i sällsynta fall.
Symtom i luftvägarna
Andra symptom som kan indikera cystisk fibros är skramlande andning och svår rastlöshet. Många barn lider av kronisk sinusinflammation. Detta märks särskilt genom smärta i ansiktet som inte kan lokaliseras exakt. Näspolyper är också vanligare hos barn med cystisk fibros än hos friska barn.
Om barn lider av andningsproblem eller matsmältningsbesvär under lång tid är det alltid lämpligt att uppsöka läkare som en försiktighetsåtgärd. Livshotande situationer kan snabbt uppstå hos barn eftersom de inte kan formulera sina klagomål själva eller inte kan bedöma deras svårighetsgrad.
Cystisk fibros: orsaker och riskfaktorer
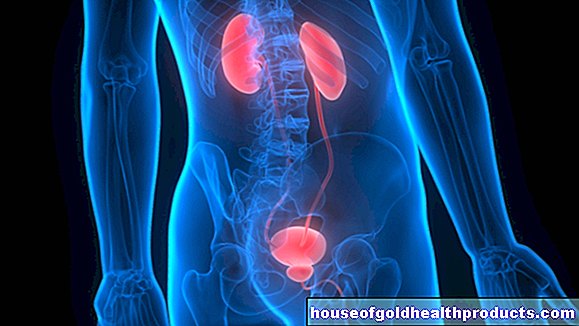
Cystisk fibros orsakas av en genetisk defekt. Den patologiska förändringen sker på den sjunde kromosomen i den så kallade CFTR-genen.
CFTR -genen (cystisk fibros transmembranregulatorgen) innehåller bygginstruktionerna för en kanal i cellmembranet genom vilket kloridjoner kommer in i cellen. De defekta kloridjonkanalerna blockerar transporten av salt till vissa kroppsceller hos patienter med cystisk fibros.
De drabbade körtelcellerna utsöndrar sedan tufft slem istället för den annars flytande utsöndringen. I lungorna, paranasala bihålor, bukspottkörteln, i tarmen, i gallvägarna och i könskörtlarna bildas hårda slemutsöndringar med högt saltinnehåll.
Cystisk fibros: hur riskerar mitt barn?
Cystisk fibros bryter bara ut när båda föräldrarna vidarebefordrar en patologiskt förändrad CFTR -gen till sitt barn. Dessa föräldrar är vanligtvis båda friska själva - de har en frisk och en defekt CFTR -gen.
Personer med cystisk fibros är endast delvis fertila. Vissa patienter blir fortfarande föräldrar. Sjuka fäder eller mödrar överför alltid en sjuk gen, eftersom båda CFTR -generna bär cystisk fibrosinformation. Dina barn blir dock bara sjuka om de också har fått en sjuk gen från den andra föräldern.
Par som har haft fall av cystisk fibros i sina familjer bör söka genetisk rådgivning innan de planerar en graviditet.
Så ärvs cystisk fibros
Preimplantationsdiagnostik
Föräldrar som kan vidarebefordra cystisk fibros till sitt barn kan dra nytta av diagnostik före implantation. Äggcellerna befruktas först artificiellt. De första celldelningarna sker i provröret (in vitro).
Innan ett embryo som skapats på detta sätt sätts in i kvinnans livmoder kontrolleras det för förändrade genetiska egenskaper.Endast embryon implanteras då som inte innehåller den sjuka genen.
Oberoende av detta kan det också undersökas under graviditeten om barnet kommer att utveckla cystisk fibros senare.
Cystisk fibros: undersökningar och diagnos
Till skillnad från för några år sedan skärmar de flesta sjukhus nu rutinmässigt efter nyfödda. Den innehåller bland annat en undersökning för cystisk fibros. Deltagande i denna screeningprocess är frivilligt och kräver samtycke från den nyfödda föräldrarna.
Screening i blod och svett
Blod tas från den nyfödda för screening. Testet för cystisk fibros har flera steg:
- Blodprov: test för ökade nivåer av immunreaktivt trypsin (IRT) och pankreatitassocierat protein (PAP). Om det finns några avvikelser utförs svettprovet.
- Svettprov: Personer med cystisk fibros har en betydligt högre salthalt i svetten än friska människor. För svettprovet cystisk fibros mäts halten av salterna natrium och klorid i kroppssvett. Hos barn samlas svett på underarmen och analyseras sedan. Om en misstanke uppstår här utförs ett genetiskt test.
Genetiskt test
Hos patienter med cystisk fibros ändras den så kallade CFTR-genen, som tillhandahåller bygginstruktioner för vissa jonkanaler. Dessa instruktioner är långa, de består av cirka 6500 baspar. Ett fel kan krypa in i koden var som helst - cystisk fibros kan därför baseras på olika genetiska förändringar (mutationer). Dessa har olika effekter. Därför testas endast de viktigaste avvikelserna i CFTR -genen.
Om båda CFTR -generna, faderns och moderns, visar en mutation, bekräftar detta - tillsammans med det positiva svett -testet - diagnosen cystisk fibros.
Familjens historia ger ledtrådar
Om ingen lämplig nyfödd screening har utförts och misstanken om cystisk fibros uppstår senare är husläkaren eller internisten rätt kontakt. I ett första samtal registrerar han den medicinska historien (anamnes). Om cystisk fibros misstänks är huvudfokus på familjens historia (finns det några kända cystiska fibros sjukdomar i familjen? Är en av föräldrarna känd för att vara bärare av en defekt CFTR -gen?).
Undersökningar
En fysisk undersökning kommer då att ske. Läkaren lyssnar på patientens lungor och skannar hans inre organ. På detta sätt kan han redan utesluta några andra sjukdomar som är associerade med symptom som liknar cystisk fibros.
Svettprovet och gentestet, som beskrivits ovan, hjälper också om cystisk fibros misstänks hos äldre barn, ungdomar eller vuxna.
När diagnosen har ställts kan läkaren använda olika undersökningar för att kontrollera i vilken utsträckning inre organ försämras i deras funktion och om komplikationer eller sekundära sjukdomar redan dyker upp. Sådana undersökningar utförs ibland rutinmässigt för att övervaka utvecklingen av cystisk fibros.
Till exempel kan blodprov användas för att avgöra om insulinutsöndringen från bukspottkörteln minskar och blodsockernivån ökar - symptom på diabetes. Leverfunktionsvärden och koncentrationen av fettlösliga vitaminer kan också mätas i blodet.
Koncentrationen av matsmältningsenzymet elastas (kommer från bukspottkörteln) och fettkoncentrationen bestäms i avföringsprover.
Vävnadsprover från patientens hals eller sputum undersöks för bakteriella infektioner i laboratoriet.
Avbildningsförfaranden (såsom röntgen, datortomografi = CT) är också ofta avslöjande. De kan till exempel användas för att upptäcka lunginfektioner. Näspolyper kan också detekteras med hjälp av CT.
Lungfunktionsmätningar (lungfunktionstester) visar hur bra lungorna fungerar.
Testar familjemedlemmar
Om cystisk fibros hittas i en familj är det vettigt att alla andra familjemedlemmar också genomgår en undersökning. Cystisk fibros kan också förekomma i milda former. Då tar det ofta många år innan cystisk fibros blir märkbar med tydliga symptom. Ändå är tidig diagnos och behandling med cystisk fibros också viktiga för dessa patienter för att öka livslängden.
Cystisk fibros: behandling
Det finns inget botemedel mot cystisk fibros. Barn födda med cystisk fibros kommer att drabbas av sjukdomens effekter resten av livet. Med en kombination av sjukgymnastik, medicinering och inandning kan sjukdomsförloppet bromsas avsevärt. Det primära målet med cystisk fibrosterapi är att göra det möjligt för de drabbade att leva ett så normalt liv som möjligt.
Lär dig leva med sjukdomen
Det är särskilt viktigt för barn att lära sig att hantera sjukdomen på lång sikt. Barn med cystisk fibros bör lära sig vad sjukdomen innebär och hur den påverkar kroppen så tidigt som möjligt. En sjukhusvistelse med specialutbildningsenheter kan vara användbar för detta. Barn och föräldrar lär sig hur de ska äta, hur sport är och hur man bäst beter sig i kritiska situationer. Föräldrar och barn informeras också om hygienåtgärder (enligt rekommendationerna från Robert Koch -institutet): regelbunden och omfattande handhygien (handtvätt med tvål och vatten), inga stoppade möbler och krukväxter.
Hjälp för lungorna
Det finns olika alternativ för att behandla symptomen på cystisk fibros. Vilka av dessa som är användbara i enskilda fall beror på patientens ålder och symptomens svårighetsgrad.
Expektoranter
Patienter med cystisk fibros lider mest av lungproblem. Regelbunden inandning med speciella tillsatser (mukolytiska medel) lossar det hårda slemmet och gör det lättare att hosta upp.
Bronkodilatatorer
Så kallade beta-2-sympatomimetika vidgar bronkierna, vilket gör andningen ännu enklare.
Läkemedel mot luftvägsinfektioner
Dålig ventilation av lungorna gör personer med cystisk fibros mer benägna att bakteriella luftvägsinfektioner. De drabbade bör behandlas med antibiotika i ett tidigt skede. Den behandlande läkaren föreskriver ofta att antibiotika ska inandas, eftersom de aktiva ingredienserna når målstället (lungorna) direkt. Alternativt eller dessutom kan intag av antibiotika (t.ex. i form av tabletter) anges.
Vid allvarliga infektioner behöver antibiotika ofta ges i venen (intravenöst). Detta kan vara nödvändigt vid till exempel kroniska infektioner med Pseudomonas aeruginosa.
Svampinfektioner i luftvägarna kan behandlas med svampdödande medel (antimykotika). Virushämmande läkemedel (antivirala läkemedel) kan ofta hjälpa till med virala luftvägsinfektioner.
Antiinflammatoriska läkemedel
Hos många patienter är luftvägarna ofta eller kroniskt inflammerade. Sedan används antiinflammatoriska läkemedel, särskilt glukokortikoider (kortison). De kan inandas, tas genom munnen eller ges i en ven.
CFTR -modulatorer
Sedan några år tillbaka finns så kallade CFTR-modulatorer för behandling av cystisk fibros. Dessa läkemedel, som tas långsiktigt, kan förbättra funktionen hos jonkanalerna som är defekta av CFTR-mutationen. Detta kan förbättra patientens lungfunktion, öka deras vikt, sänka saltinnehållet i svett och minska frekvensen av lunginfektioner.
Detta är emellertid mutationsspecifika terapier, vilket innebär: CFTR-modulatorer fungerar bara med vissa mutationer och därför bara för vissa patienter. Några exempel:
Ivacaftor godkändes som den första CFTR -modulatorn 2012. Det kan ordineras till spädbarn från 12 månader och som väger minst sju kilo om de har minst en så kallad gating-mutation (t.ex. G551D).
Cystisk fibrospatienter 12 år och äldre som har två kopior av F508del -mutationen kan behandlas med en kombination av de två CFTR -modulatorerna ivakaftor och lumakaftor.
Forskare arbetar intensivt med andra läkemedel som hjälper mot andra mutationer som ligger till grund för cystisk fibros.
Fysioterapier
Olika fysioterapitekniker hjälper till att befria luftvägarna från tuffa bronkiala sekret. Dessa inkluderar dräneringslagring, vibrationsdränering, tappmassage och hostträning. Föräldrar till små barn med cystisk fibros kan lära sig dessa andningsterapitekniker och använda dem dagligen hemma. Äldre barn och vuxna med cystisk fibros kan utföra andningsterapi självständigt (efter instruktion från en terapeut).
Lungtransplantation - det sista hoppet
Om cystisk fibros är allvarlig är en lungtransplantation ett alternativ. Så drastiskt som det här steget kan verka från början kan många patienter sedan leva ett liv med betydligt färre symtom.
Äter ordentligt för cystisk fibros
Eftersom matsmältningen störs vid cystisk fibros måste patienterna vara noga med sin kost. Måltider bör ge tillräckligt med kalorier och protein för att stödja normal tillväxt. Ett normalt till högt fettintag rekommenderas också. Patienter får matsmältningsenzymer för att stödja matutnyttjandet. Det finns också preparat med fettlösliga vitaminer (A, D, E, K) och mineraler. Det senare ersätter de salter som patienter svettas ut i stora mängder.
En viss mängd bordsalt bör intas med mat hela dagen. Motsvarande mer krävs vid höga temperaturer, feber, diarré, kräkningar. En eventuell järnbrist bör testas en gång om året och ersättas vid brist.
Spädbarn med cystisk fibros
Spädbarn med cystisk fibros bör endast matas med bröstmjölk under de första fyra månaderna, så länge de trivs. Det finns till och med bevis för att amning i mer än sex månader är förknippat med mindre svårighetsgrad. Forskning har visat att ammande barn har bättre lungfunktion och färre infektioner under de tre första levnadsåren än spädbarn med cystisk fibros som får modersmjölksersättning.
Om bröstmjölksmatning inte är möjlig bör spädbarn ges kommersiell formel när de trivs.
Om spädbarn med cystisk fibros inte frodas rekommenderas högkaloririka för barn (förutom bröstmjölk för ammande barn).
När det gäller introduktion av kompletterande mat gäller samma rekommendationer som för friska barn: föräldrar bör börja komplettera matning tidigast i början av den femte månaden och senast i början av den sjunde månaden.
Kosten för barn med cystisk fibros bör anpassas efter deras behov så att de små kan trivas efter deras ålder. Både överviktiga och underviktiga bör undvikas. En kost av god kvalitet är också tillrådligt. Du bör vara uppmärksam på bra kostfetter (med enkelomättade och fleromättade fettsyror, omega-3-fettsyror).
Lungfunktionen eller alveolernas tillväxt påverkas av fysisk utveckling under de första åren av livet. Därför bör barn med cystisk fibros i denna åldersgrupp regelbundet undersökas för näringsstatus (baserat på mätningar av vikt, kroppslängd och huvudomkrets).
Ta vaccinationer
Vaccinationer är särskilt viktiga för personer med cystisk fibros. Bakterier har lättare med dem, och de är ofta mer sjuka än patienter som inte är stressade på förhand.
I grund och botten bör barn med cystisk fibros få alla vaccinationer som ständiga vaccinationskommissionen (STIKO) rekommenderar för friska avkommor. Dessutom rekommenderas en hepatit A -vaccination för barn i åldern 12 månader och uppåt, och en årlig influensavaccination med tetravalent inaktiverat influensavaccin för barn i sex månader och uppåt. För spädbarn under sex månader bör alla hushållsmedlemmar och vårdgivare vaccineras mot influensa för att skydda barnet.
Barn med cystisk fibros kan också ges ett förebyggande läkemedel (palivizumab) mot RSV -patogenen (respiratoriskt syncytialvirus). Dessa virus, som sprids över hela världen, kan orsaka luftvägssjukdomar, särskilt hos spädbarn och småbarn.
Cystisk fibros: sjukdomsförlopp och prognos
Cystisk fibros orsakas av en förändring i det genetiska sminket och är därför obotligt. I allmänhet minskar livslängden och livskvaliteten vanligtvis signifikant vid cystisk fibros. Utan terapi försämras hälsotillståndet snabbt och de drabbade lever vanligtvis inte länge.
Med snabb och konsekvent behandling kan sjukdomsförloppet bromsas avsevärt. Patienterna lever nu betydligt längre än för några år sedan. Medellivslängden med cystisk fibros är för närvarande cirka 50 år. Men många patienter blir också äldre.
Komplikationer och sekundära sjukdomar
Även med intensiv terapi kan komplikationer uppstå igen och igen med cystisk fibros. Oftast uppstår akut andfåddhet på grund av dålig lungventilation. Enskilda områden i lungorna kan till och med kollapsa (atelektas).
Kronisk bronkit eller lunginflammation utvecklas ofta. Svampar kan också påverka lungorna.
Dessutom kan förändringar i vätske- och elektrolytbalansen utlösa chock och cirkulationssvikt.
Andra möjliga komplikationer och sekundära sjukdomar vid cystisk fibros är:
- kronisk leversjukdom, särskilt levercirros
- Inflammation i gallblåsan och gallsten
- kronisk pankreatit
- störd hjärtfunktion
- akut tarmobstruktion (ileus)
- Intestinal invagination (intussusception)
- Undernäring
- Diabetes mellitus
- minskad fertilitet hos kvinnor och infertilitet hos män
Cystisk fibros: förebyggande?
Cystisk fibros är en ärftlig sjukdom, så förebyggande är inte möjligt. Personer med ökad familjerisk bör söka genetisk rådgivning om de vill skaffa barn. Ett genetiskt test utförs för att avgöra om CFTR -genen har ändrats. Beroende på om en eller båda parter bär den defekta genen kan risken för avkomman beräknas.
Under tiden är diagnos före implantation (PGD) möjlig för cystisk fibros i de flesta europeiska länder enligt lagens bestämmelser i respektive land. Äggcellerna befruktas utanför livmodern och endast embryon utan de problematiska cystiska fibrosgenerna används. Förutsättningen för detta är alltid godkännande av en etisk kommitté.
Ytterligare information
Riktlinjer:
- S2 konsensusriktlinje "Diagnos av cystisk fibros" från Society for Pediatric Pneumology
- S3 riktlinje "Lungsjukdom vid cystisk fibros" från Society for Pediatric Pneumology
- S3 -riktlinje: "Cystisk fibros hos barn under de två första levnadsåren, diagnostik och terapi" från Society for Pediatric Pneumology (GPP), German Society for Child and Adolescent Medicine (DGKJ) m.fl.
Självhjälpsgrupper:
- Cystisk fibros e.V.: Https://www.muko.info/
- CF Österrike: Cystisk fibros Hjälp Österrike: https://www.cf-austria.at/
- Cystisk fibros Schweiz: https://cystischefibroseschweiz.ch/







.jpg)






.jpg)