Amyotrofisk lateral skleros
Sophie Matzik är frilansskribent för medicinska team.
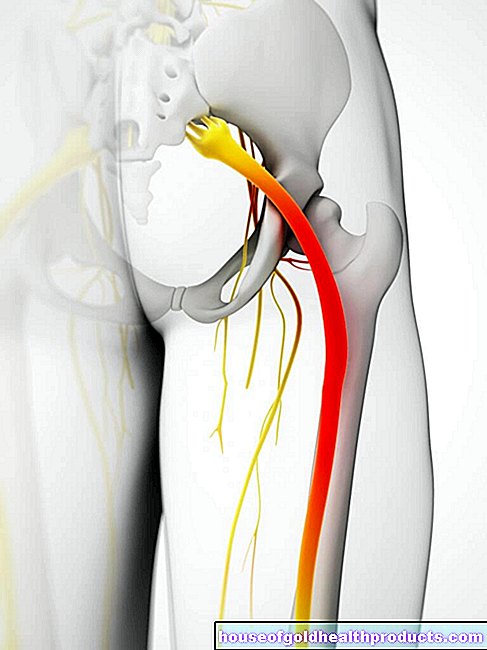
Mer om -experterna Allt -innehåll kontrolleras av medicinska journalister.Amyotrofisk lateral skleros (ALS) är en kronisk degenerativ sjukdom i centrala nervsystemet. Patienterna lider av progressiv muskelförlamning. De måste tidigt förlita sig på en rullstol, och senare har de svårt att svälja, tala eller andas. I många fall leder ALS till döden inom några år. Men det finns också människor som astrofysikern Steven Hawking, som levt med sjukdomen i årtionden. Läs mer om tecken och behandling av amyotrofisk lateral skleros här.
ICD -koder för denna sjukdom: ICD -koder är internationellt erkända koder för medicinska diagnoser. De finns till exempel i läkarbrev eller på intyg om arbetsoförmåga. G12
Amyotrofisk lateral skleros: beskrivning
Amyotrofisk lateral skleros (även känd som ALS, myatrofisk lateral skleros, Charcots sjukdom eller Lou Gehrigs syndrom) har varit känd som en nervös sjukdom sedan mitten av 1800-talet. Jämfört med andra neurodegenerativa sjukdomar förekommer det relativt sällan; experter uppskattar att cirka två av 100 000 människor utvecklar amyotrofisk lateral skleros varje år. Antalet nya fall av amyotrofisk lateral skleros är högre i Europa än i andra delar av världen.
I de flesta fall inträffar amyotrofisk lateral skleros mellan 50 och 70 år. Det börjar vanligtvis med muskelsvaghet och muskelsvinn i extremiteterna. I den senare processen kommer ansiktsmusklerna attrofi (atrofi). Detta kan åtföljas av fronto-temporal demens. Ofrivilliga kramper i extremiteterna, så kallad spasticitet, är också en del av amyotrofisk lateral skleros. .
De tre formerna av amyotrofisk lateral skleros
Om amyotrofisk lateral skleros diagnostiseras under den medicinska undersökningen, skiljer specialisten mellan tre former:
- Sporadisk form: amyotrofisk lateral skleros har ingen identifierbar orsak.
- Familjform: Amyotrofisk lateral skleros utlöses av en förändring av den genetiska informationen om en kromosom (genetisk mutation) som överförs från föräldrar till deras barn.
- Endemisk form: Orsaken till ALS -sjukdom har ännu inte klargjorts på vissa områden.
Vad händer med amyotrofisk lateral skleros
Amyotrofisk lateral skleros är en nervsjukdom. Nervcellerna (neuroner) som är ansvariga för att överföra kommandon till musklerna försvinner gradvis när sjukdomen fortskrider - en process som inte längre kan vändas. Kommandona från hjärnan och ryggmärgen blir svagare och svagare och överförs senare till musklerna. Amyotrofisk lateral skleros manifesterar sig därför först som muskelsvaghet, senare uppstår symtom på förlamning.
Hos de flesta patienter har amyotrofisk lateral skleros inga negativa effekter på de mentala förmågorna: sensorisk uppfattning, medvetande och tänkande förmåga förblir helt intakt. Endast cirka fem procent lider av minnesförlust. Det är desto mer tragiskt för de drabbade när de inte längre klarar vardagen ensam under ALS -sjukdomen. Amyotrofisk lateral skleros är obotlig och de drabbade dör vanligtvis av konsekvenserna inom bara några år.
Amyotrofisk lateral skleros: symptom
Den kliniska bilden av ALS kan skilja sig avsevärt från person till person. 25 procent av de drabbade klagar initialt på muskelsvaghet och muskelsvinn i de små handmusklerna och senare i armmusklerna. Amyotrofisk lateral skleros kännetecknas också av kramper i benen och en nedgång i ansiktsmusklerna. Symtom kan också initialt kännas i fötterna och benen och först senare spridas till armarna och ansiktsmusklerna.
Slutligen inkluderar förloppet av ALS -sjukdomen också en minskning av ansiktsuttryck. Det orsakas av atrofi i ansiktsmusklerna och hindrar också förmågan att tala. Tal blir alltmer långsamt och suddigt (glödlampsspråk), och svälja kan också vara svårt när amyotrofisk lateral skleros fortskrider.
Hos cirka 20 procent av personer med Lou Gehrig syndrom (ALS -sjukdom) dör nervcellerna i ett visst område av hjärnan först. De första märkbara symtomen är en minskning av talförmågan och sväljstörningar (bulbar förlamning). Först senare minskar muskelstyrka och volym i extremiteterna.
Andra symtom på amyotrofisk lateral skleros kan innefatta ofrivilliga rörelser, fascikulationer (okoordinerade ryckningar i enskilda muskelknippor), brist på muskelreflexer och okontrollerat skratt och gråt som ofta förekommer. I de senare stadierna kännetecknas alla former av ALS av svåra kramper och förlamning.
Amyotrofisk lateral skleros: orsaker och riskfaktorer
Amyotrofisk lateral skleros har länge varit känd som en nervös sjukdom, men orsakerna till sjukdomen är fortfarande i stort sett oförklarliga. Även om en viss frekvens observeras i familjer med en predisposition, uppstår majoriteten av fallen av sjukdomen spontant. Var hundradels person med ALS har en ärftlig eller nyuppträtt mutation i en viss gen som är viktig för cellmetabolismen. Det stör bildandet av ett enzym som avlägsnar fria syreradikaler i cellen. Om det misslyckas vid amyotrofisk lateral skleros kan dessa radikaler skada nervcellerna avsevärt. Detta stör överföringen av kommandon till musklerna - resultatet är muskelsvaghet och slutligen fullständig förlamning.
Amyotrofisk lateral skleros: undersökningar och diagnos
ALS -diagnosen bekräftas av olika medicinska undersökningar. Några av dessa är obligatoriska; den behandlande läkaren kan använda ytterligare tester för att utesluta andra sjukdomar med symtom som liknar amyotrofisk lateral skleros (differentialdiagnos).
Typiska undersökningar för amyotrofisk lateral skleros är:
- Test av nervfunktioner (reflexer, muskelspänningar, beröringskänsla, urinblåsa och tarmkontroll)
- Test av psykologisk och mental prestation
- Elektromyografi och neurografi (registrering av elektrisk muskelaktivitet)
- Blodprov
- Test av lungfunktion, möjligen även en blodgasanalys
- Magnetic resonance imaging (för att skilja ALS -sjukdom från andra nervsjukdomar)
- Bestämning av kroppsvikt och kroppsmassindex (BMI)
En ALS -diagnos kan också bekräftas ytterligare med följande tester:
- Muskelbiopsi (struktur av muskelfibrer)
- Test för demens
- Magnetisk stimulering av motorhjärnans områden (transkraniell magnetisk stimulering)
- Undersökning av blod och sprit för olika patogener (Borrelia, HIV, Lues, etc.) eller antikroppar
- ENT -undersökning (differentialdiagnos: tal- och sväljstörningar)
- Andra avbildningsförfaranden (t.ex. magnetisk resonansavbildning av hjärnan och ryggmärgen)
- Undersökning av blodserumet för metaboliska störningar
- Benmärgsbiopsi
- Registrering av näringsstatus (oberoende förutsägare för förväntad livslängd)
På grund av de höga kostnaderna är ett DNA-test för en förändrad SOD-1-gen bara meningsfullt om familjen till den drabbade personen redan har utvecklat amyotrofisk lateral skleros. Det är avgörande för diagnosen ALS att utesluta underformer som primär lateral skleros eller Kennedys syndrom samt överlappningar med andra neurologiska sjukdomar (t.ex. Parkinsons sjukdom).
Om diagnosen ALS / Lou Gehrig syndrom har ställts av en läkare kan det vara meningsfullt att söka yttrande från en annan expert på grund av dess enorma betydelse.
Amyotrofisk lateral skleros: behandling
Amyotrofisk lateral skleros (ALS) är obotlig. Målet med all behandling för ALS -sjukdom är därför att göra det möjligt för de drabbade att leva så länge som möjligt, självständigt och med få symtom. Patienter får den bästa vården på en klinik som har ett tvärvetenskapligt team tillgängligt för behandling av amyotrofisk lateral skleros. Förutom neurologer omfattar detta även gastrointestinala specialister (gastroenterologer), pulmonologer (pulmonologer), logopeder (logopeder), psykologer, sjukgymnaster och arbetsterapeuter, tandläkare och dietister.
Den aktiva ingrediensen riluzol är för närvarande tillgänglig som den enda rekommenderade läkemedelsbehandlingen för amyotrofisk lateral skleros. Även om det inte tillåter läkning, kan det skydda nervcellerna från ytterligare skador genom att hämma neurotransmittorn glutamat i hjärnan (glutamatantagonist). Riluzole är därför att förstå som ett läkemedel som bromsar sjukdomsförloppet. Ju tidigare terapi börjar, desto mer minskar symtomen på amyotrofisk lateral skleros. Medellivslängd och livskvalitet ökar avsevärt till följd av läkemedlet. Ytterligare aktiva ingredienser testas för närvarande kliniskt och kommer, om de lyckas, att godkännas under de närmaste åren.
Amyotrofisk lateral skleros: symptom kan behandlas
Eftersom amyotrofisk lateral skleros är väldigt individuell kan de drabbade drabbas av mycket olika symptom som behandlas individuellt. Många patienter får andningsproblem och lider av andfåddhet eftersom andningsmusklerna inte längre fungerar korrekt. En hemmaventilator och mukolytiska läkemedel kan hjälpa. Förebyggande åtgärder mot lunginflammation är också ofta användbara.
Problem med att svälja kan göra konstgjord matning nödvändig. Detta görs antingen via infusioner (via venen) eller via ett PEG -rör som placeras genom bukväggen direkt in i magen. Ökad salivation (hypersalivation) är inte heller ovanligt under ALS -sjukdomen. För behandling används läkemedel som dämpar påverkan av det parasympatiska nervsystemet - som amitriptylin, skopolamin eller metionin.
Sjukgymnastik och arbetsterapi kan också hjälpa till med amyotrofisk lateral skleros. Symtom som okontrollerad muskelryckning eller risken för att blodproppar bildas i benvenerna (trombos) kan därmed minskas. Smärta kan ofta hanteras väl med starka smärtstillande medel (opioider) och andra smärtstillande medel. För behandling av depression och panikattacker stöds psykoterapi med lämpliga läkemedel (psykofarmaka).
De drabbade har stora förhoppningar om stamcellsterapi. Dessa ännu icke differentierade kroppsceller ska ersätta några av de nervceller som har dött. Cellerna tas från patientens blod eller benmärg, odlas och implanteras slutligen i de skadade områdena. En slutlig bedömning av denna behandling är för närvarande inte möjlig trots några positiva resultat. Dessutom är denna behandlingsform inte tillåten i Tyskland.
Amyotrofisk lateral skleros: sjukdomsförlopp och prognos
Livslängden för personer med amyotrofisk lateral skleros (ALS) kan knappast förutsägas på grund av sjukdomens olika förlopp. Den genomsnittliga livslängden från diagnostiden är tre och ett halvt år, men en tredjedel av patienterna lever fortfarande fem år eller mer.
Om ALS -sjukdomen diagnostiseras, brukar läkare råda tidigt, på grund av den ofta fulminanta sjukdomsförloppet, att utfärda ett levande testamente och en vårdproxy och att söka psykologiskt stöd. Förutom läkare erbjuder ALS självhjälpsgrupper också råd och hjälp.
Om diagnosen amyotrofisk lateral skleros ställs tidigt och behandlingen påbörjas omedelbart är en mycket hög livskvalitet möjlig för många patienter. Symtom som depression och ångest är sällsynta vid ALS -sjukdom. Om god vård säkerställs i hemmiljön är sluten behandling på kliniken endast nödvändig i den sena fasen av amyotrofisk lateral skleros.
Tagg: anatomi förebyggande sjukhus


-mit-tattoos-dem-schicksal-trotzen.jpg)